Introduction
With the current trend in the democratisation of massive parallel sequencing, several laboratories throughout the world invest important efforts in deciphering the three-dimensional chromatin organisation, mainly by using proximity ligation approaches. Indeed, since the development of the Hi-C assay by Lieberman-Aiden et al. in 2009 (Science 2009), more than 140 related publications are indexed in Medline (June 2015) and more than 300 datasets have been deposited in the public repository Gene Expression Omnibus (GEO). However, we are still missing quantitative methods
- to evaluate the overall quality of each of these publicly available datasets and
- to explore their content in a user-friendly manner.
To address these issues, we have developed LOGIQA (Long-range Genome Interactions Quality Assessment), a database hosting quality scores for long-range chromatin interaction assays. Below we describe the various steps applied to assess the local and global quality scores, as well as the multiple options available for exploring the content of this database.
Data processing
As part of the NGS-QC Generator project, aiming at qualifying ChIP-sequencing and enrichment-related datasets available in the public domain, we have developed an automated pipeline able to (i) download public datasets, (ii) align them to the corresponding reference genome, (iii) infer quality indicators by the NGS-QC Generator procedure and finally (iv) compile them into the dedicated database. Following the same strategy, we have added a module handling data coming from Hi-C and related proximity-mediated ligation assays.
Our pipeline is using several widely used, freely available tools. Briefly, we follow the steps described bellow.
Read mapping
The latest samples released on the Gene Expression Omnibus are selected and added to the pipeline's queue.
Raw sequencing data are downloaded from the Sequence Read Archive, then converted to fastq format using SRA toolkit's fastq-dump.
Reads are aligned using using bowtie2 against the dm3 build (fruitfly), hg19 build (human), or mm9 (mouse).
We are using the --very-sensitive-local preset, in order to filter low quality and adapter-contaminated reads. Only uniquely mapped reads are kept.
For Hi-C and ChIA-PET data, paired-end reads are aligned separately.
Read filtering (Hi-C/ChIA-PET)
After mapping paired-end reads, we remove PCR duplicates. PCR duplicates are defined as read pairs sharing the same mapping position and orientation.
Finally, we remove inter-chromosomal interactions, as well as intra-chromosomal interactions shorted than 10 kb from the remaining pairs.
Binning
We first establish three substets by randomly selecting 90%, 70%, and 50% of the retained reads. Then, we bin the data by assigning reads to fixed-size genomic intervals (5kb and 25 kb for Hi-C/ChIA-PET, 500 bp for 4C-seq).
Quality assessment
Hi-C/ChIA-PET
LOGIQA is based on the principles applied by the NGS-QC Generator tool for assessing quality descriptors. Basically we use random sampling to create subsets of the original dataset, which are then used to evaluate the changes in enrichment patterns (“robustness”) as a consequence of this treatment. The underlying hypothesis is that, under optimal conditions, the enrichment patterns retrieved from the random sampling subsets should differ only by the absolute amplitude relative to the original dataset. Importantly, the enrichment levels are expected to change proportionally to the percentage of random sub-sampling (i.e., 90% of the original enrichment levels when 90% of the initial reads were used for random sampling). We infer numerical quality indicators for given global profile or a locally defined region by measuring the deviation from the theoretically expected enrichment.
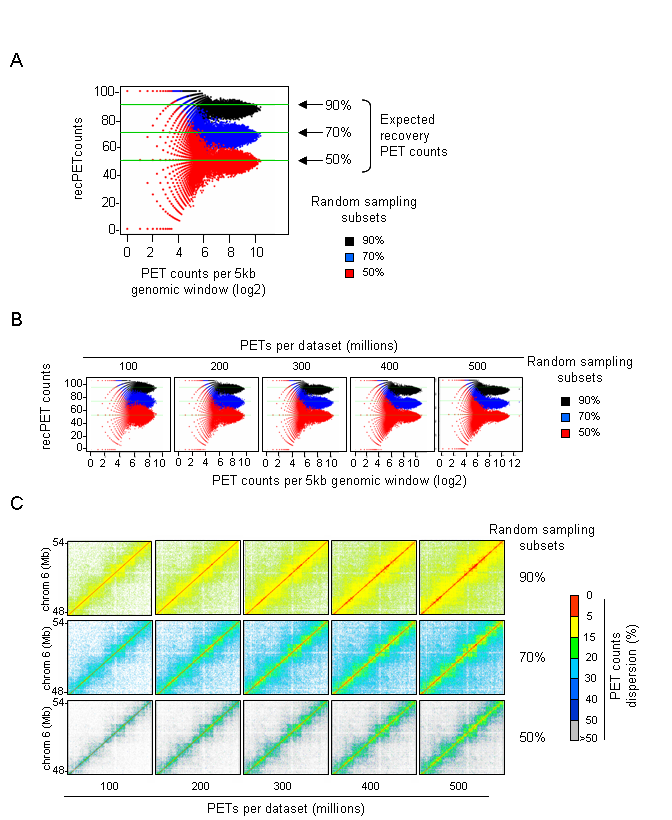
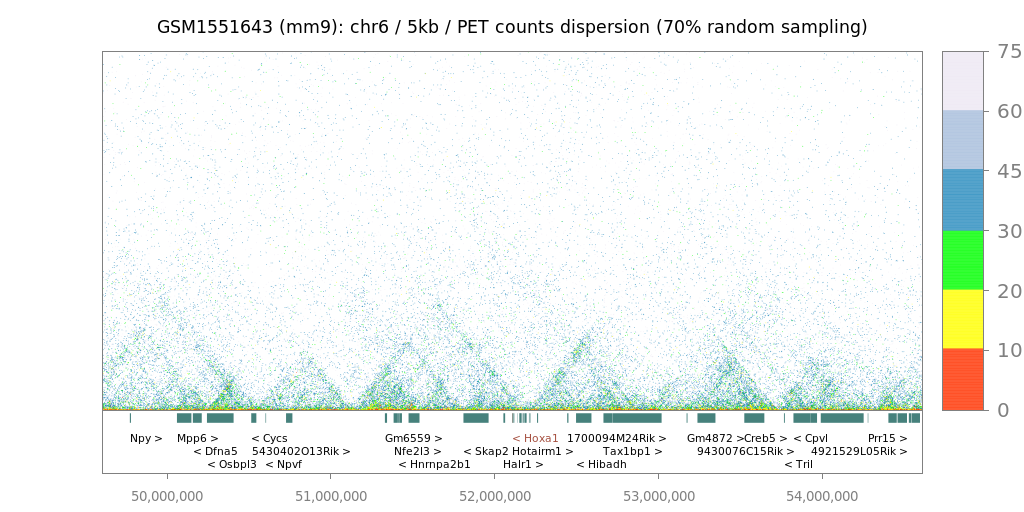
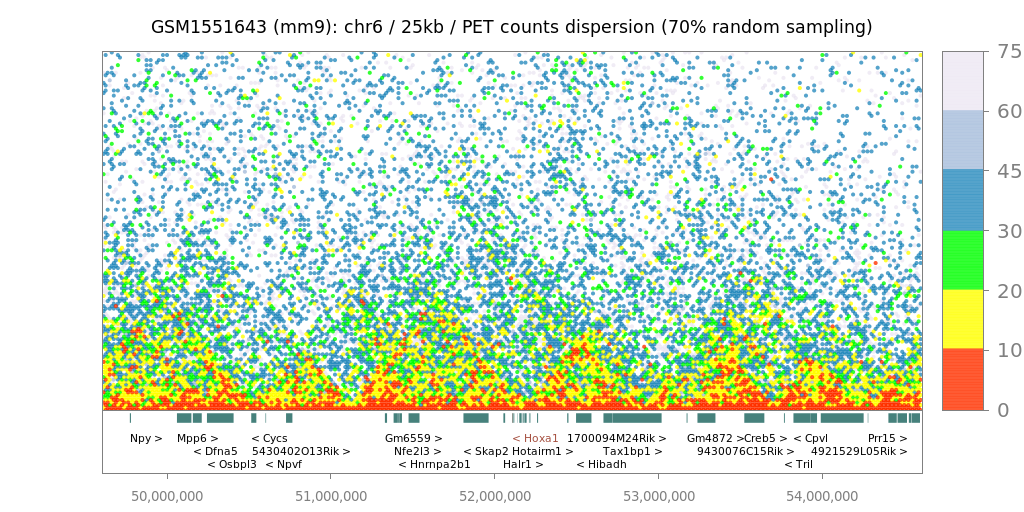
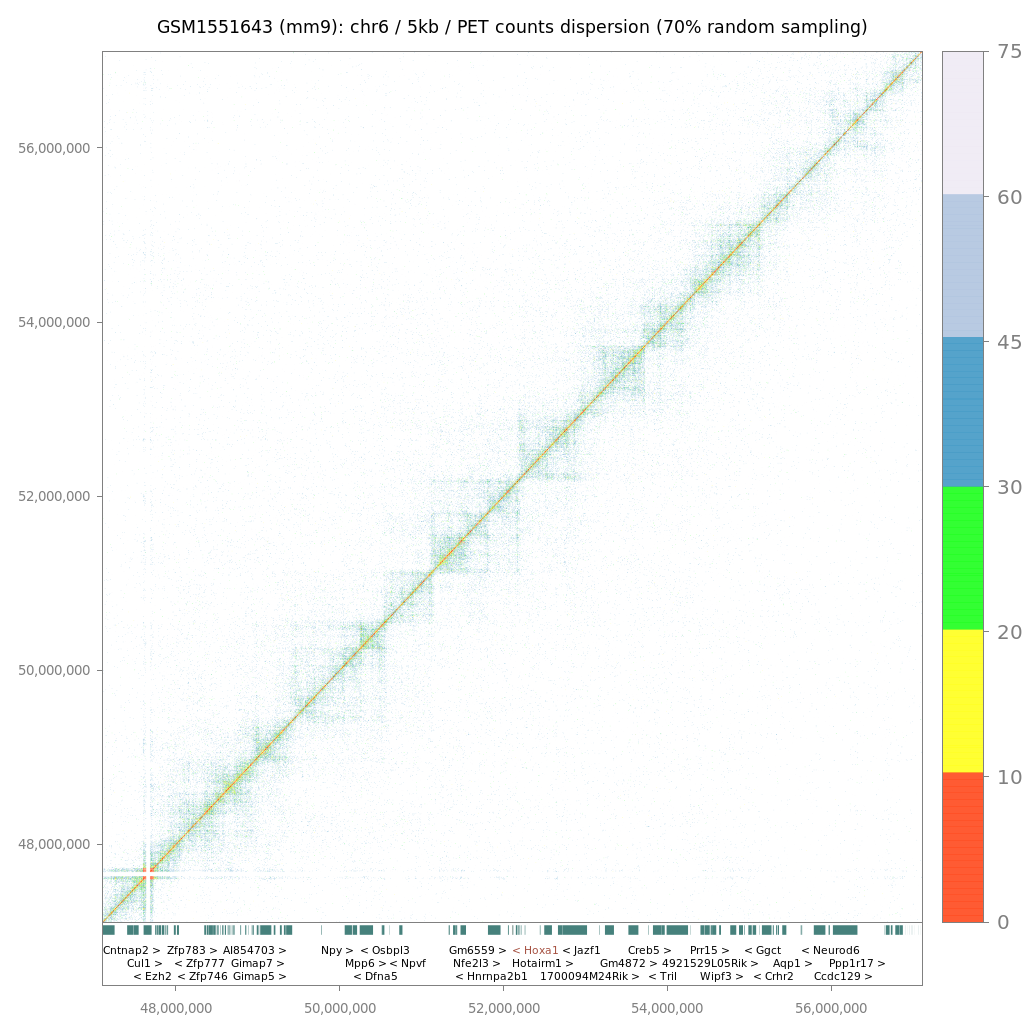
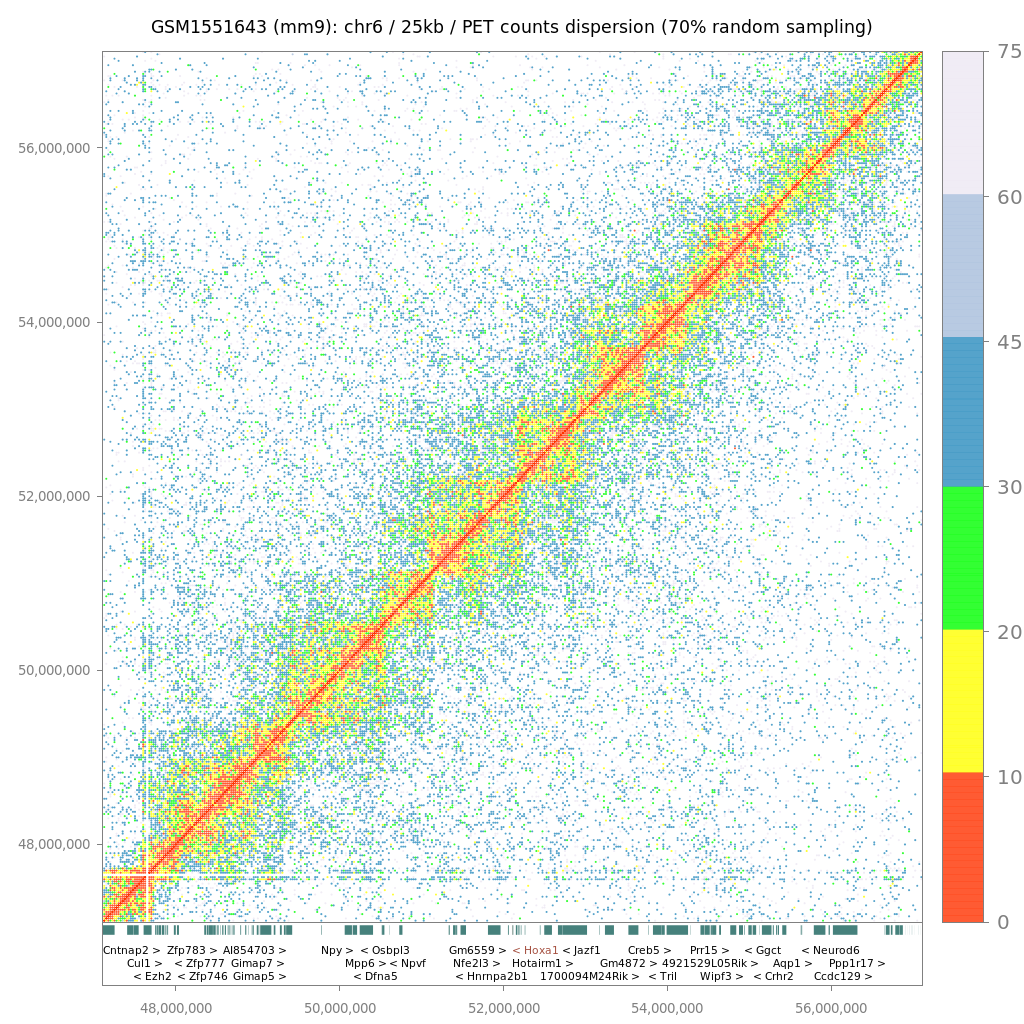
Since LOGIQA is designed for quality assessment of long-range interaction assays, random sampling is performed from the paired-end reads or tags (PETs), which are used for annotating interaction between distal genomic regions. As illustrated in figure i, the first step consists of establishing random sampling subsets (90%, 70%, 50%; described herein as s90, s70 or s50), as previously done by the NGS-QC Generator. For this we consider only PETs that describe intra-chromosomal interactions spanning a distance longer than 10kb.

After random sampling, intra-chromosomal interaction maps are reconstructed by assessing the number of PET counts within either 5 kb or 25 kb regions. Importantly, these two different windows of resolution provide the option of a rather high resolution (for future comparison with other types of datasets) and represent a uniform way to analyze variable types of datasets; this concerns particularly Hi-C assays performed with different restriction enzymes or ChIA-PET assays performed with sonication-sheared chromatin.
The quality assessment is performed by comparing the PET counts per defined genomic window in the random sampling subsets relative to that observed in the original dataset. This is performed by the following equation:
$$recPETcounts={samPETcounts}/{oPETcounts}*100$$where samPETcounts correspond to PET counts assessed after random sampling and oPETcounts correspond to those retrieved with the original dataset.
As illustrated in the scatter-plot in figure iia, genomic windows with high PET counts show a recovery
of the expected proportional PET counts (recPETcounts); i.e. 90%, 70% or 50% for s90, s70 or s50 subsets respectively.
It is also evident that a decrease in PET counts correlates with an increase of the deviation from the expected proportional behavior.
Importantly, taking advantage of a large Hi-C dataset that contains more than 600 million sequenced PETs,
we have established subsequent subsets (100, 200, 300, 400 and 500 million PETs), which were used as a quality calibration scale system.
The quality calibration scale system has been set up by merging two Hi-C datasets to obtain a dense meta-dataset.
Importantly, the use of this calibration system allowed to evidence an overall gain in similarity between the observed recPETcounts and the expected proportional behavior (figure iib). This behavior can be quantified by measuring the observed recPETcounts dispersion levels as following:
$$δPETcounts = samd - recPETcounts$$where δPETcounts correspond to the PET counts dispersion and samd to the random sampling density (90%, 70% or 50%). In this manner, each evaluated genomic region (5kb or 25kb window) can be expressed by its δPETcounts assessed for a given random sampling subset analysis, as illustrated in figure iic. Importantly, representing genome interaction maps in the context of PET counts dispersion (δPETcounts) transforms the display into a uniform scale for comparing datasets generated at variable PET sequencing levels (e.g. PET count dispersion: 5-50%).
Finally, while δPETcounts interaction maps provide a visual display of the quality associated to a given genomic region, they do not allow evaluation of the quality of the entire dataset. Therefore, we defined the following two global quality descriptors:
- Density quality indicators (denQCi): the fraction of genomic regions (5kb or 25kb window) in the random sampled datasets presenting δPETcounts lower than a defined threshold. Specifically, LOGIQA presents denQCi values computed for 90%, 70% and 50% random samplings (denQC.90, denQC.70 and denQC.50 respectively).
- Similarity quality indicators (simQCi): the ratio between two denQCis is used to evaluate their degree of similarity. Specifically, LOGIQA presents simQCi values computed for denQC.90 and denQC.70 relative to denQC.50 (simQC.90/50 and simQC.70/50 respectively).
Note that denQCi aims at quantifying the proportion of genomic regions presenting PET count dispersion levels for a given random sampling. In fact, an s90 random sampling presents generally less variation from the original dataset, while the s50 subset will have the highest deviation. The simQCi measures the relative difference between denQC indicators; consequently, high quality datasets have simQCi values closer to 1 (no difference between the compared denQCis) and the highest possible levels for the denQC.50.
Finally, we defined QCscore, which combines the previous metrics (denQCi and simQCi) according to the following formula:
$$QCscore={denQC.50}/{{simQC.90}\/50}*{denQC.50}/{{simQC.70}\/50}$$The QCscore provides a quality readout, in which the influence of both the denQC.50 and the simQCis computed for s90 relative to s50 (simQC.90/50), and s70 relative to s50 (simQC.70/s50) are represented.
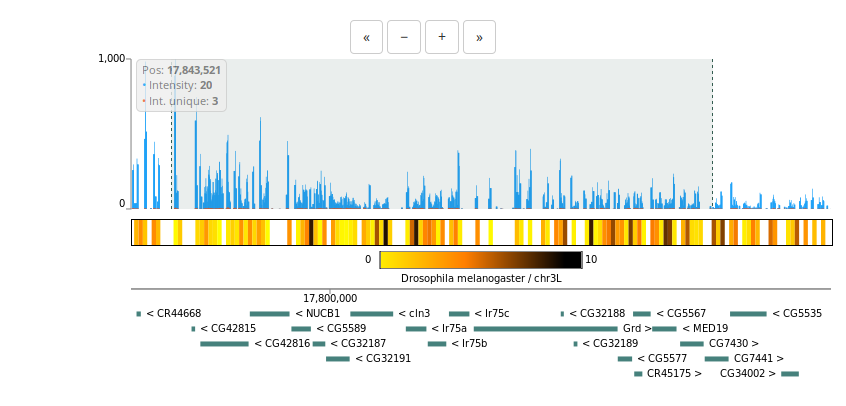
4C-seq
For 4C-seq, we are using the standard NGS-QC Generator. For more information, please refer to the NGS-QC Generator documentation.Data exploration
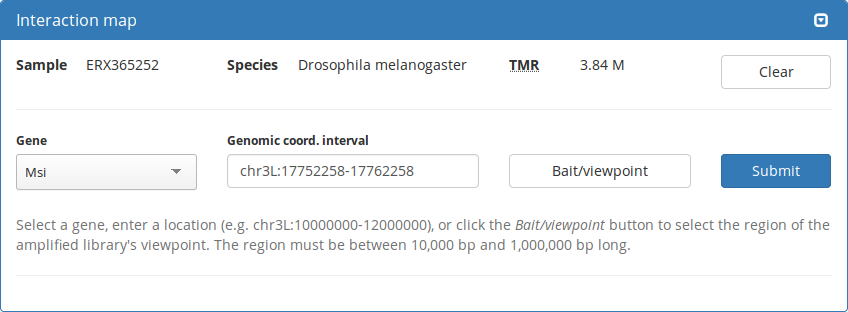
LOGIQA is a user-friendly, single-page web application with filtering, searching, sorting, and data visualization features.

Global quality plot
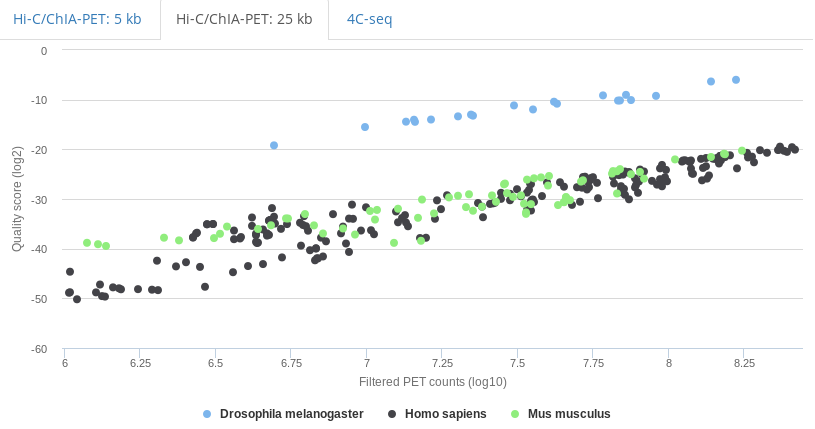
Experiments table
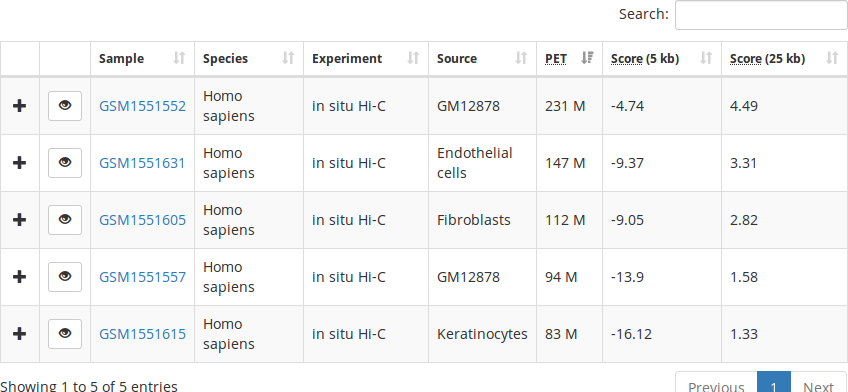
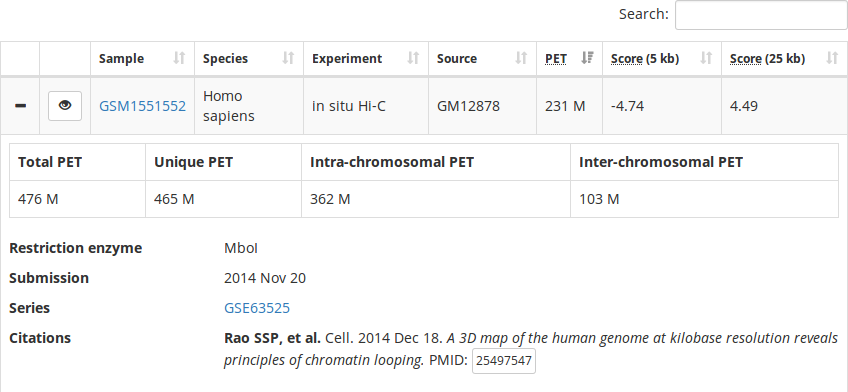
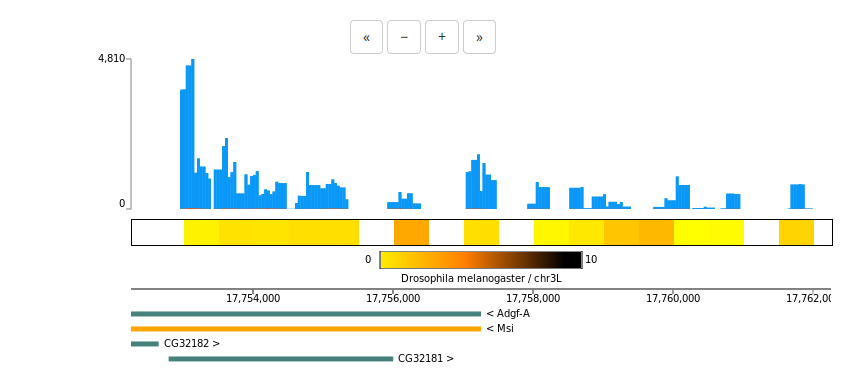
Interaction maps
Hi-C/ChIA-PET

4C-seq